The Modern Era of Medical Genetics: Three Examples of “Excellent Knowledge”
From our contemporary perspective, the work and insights of Garrod are landmarks. There can be no doubt that the incorporation of the Mendelian laws ofheredity into the study of human diseases was the turning point in the history of medical genetics.
Surprisingly, however, Garrod’s theories and suggestions went largely unnoticed for several decades. One reason for this slow recognition, according to H. Harris (1963), was that it was quite impossible at the time Garrod was writing for most geneticists to reduce Mendel’s laws to purely chemical phenomena. Indeed, in the earliest days of the rediscovery of Mendel much of the effort being expended in formal genetics was devoted to coming to terms with a growing list of exceptions to those laws (Carlson 1966).This is not to say that medical genetics had been put on hold until the middle of the twentieth century. In fact, progress in Lmderstanding the hereditary nature of numerous human diseases was being made, but it did not truly come together until the middle of this century. Two disorders that contributed greatly to this progress were sickle-cell anemia and Down syndrome.
The Triumph of the Molecular Model: SickleCell Anemia
G. J. Brewer (1985) has commented that sickle cell anemia (14190) is one of the most fascinating diseases in medicine. And this one disease may be credited with having ushered in the era of molecular medicine.
The first case of sickle-cell anemia was reported in 1910 by a Chicago physician, J. B. Herrick. Upon his examination of a young black man from Grenada, West Indies, he found, among other features, “a secondary anemia not remarkable for the great reduction in red corpuscles or hemoglobin, but strikingly atypical in the large number of nucleated red corpuscles of the normoblastic type and in the tendency of the erythrocytes to assume a slender sickle-like shape” (Herrick 1910).
Herrick had never before encountered a similar abnormality and reported:Whether the blood picture represents merely a freakish Poikilocytosis or is dependent on some peculiar physical or chemical condition of the blood, or is characteristic of some particular disease, I cannot at present answer. I report some details that may seem non-essential, thinking that if a similar blood condition is found in some other case a comparison of clinical conditions may help in solving the problem. (Herrick 1910)
Soon after, a similar case was reported, and within a very few years, the first fully scientific investigation of the sickling phenomenon was described. That study, published in 1917 by V. E. Emmel, was based on the case of a young black woman in St. Louis. The patient had an ulceration on her leg and severe anemia, and “instead of the typical rounded disk form, about one-third of the corpuscles are greatly elongated in shape. A large percentage of the latter have a rounded, rod-like shape with more or less tapered ends, and as a rule present a curved or crescentic form” (Emmel 1917). Emmel was the first to show that the sickling of the red cells was developmental, with seemingly normal, enucleate red cells undergoing the sickling change as they matured. He further showed that none of the red cells from controls or from any other type of anemia or leukemia could be induced to sickle. However, Emmel did find that a small proportion of the red cells from the patient’s father, who was not anemic, did undergo sickling in cell culture conditions.
Curiously, even though the father of the patient showed traces of the sickling trait and three of the patient’s siblings had died at an early age of severe anemia, Emmel made no reference to a possible hereditary component in the disorder. The first such suggestion was offered six years later by J. G. Huck (1923). In addition to reporting the first large clinical sample, 17 patients, Huck displayed the pedigrees of two sickle-cell families.
In one of these families, both parents were affected but had produced one normal offspring in addition to two affected offspring. The significance of these families was noted: “Apparently the ‘sickle cell’ condition in man is inherited according to the Mendelian law for the inheritance of a single factor.” Moreover, “one interesting feature of this inheritance is the fact that the sickle cell condition is dominant over the normal condition” (Huck 1923). Over the course of 13 years, reports of a unique pathological finding had led to the recognition of a hereditary disease - sickle-cell anemia.After this good start, the study of sickle-cell anemia slowed dramatically - partly because most investigators failed to recognize a fundamental difference between sickle-cell anemia and the nonanemic sickle-cell trait. Most believed that the latter was merely the dormant stage of the former. In 1933 L. W. Diggs and his colleagues suggested that this interpretation was incorrect. They showed that the percentages ofhemoglobin determinations among black schoolchildren with the sickle-cell trait were not different from those of controls. In addition, no significant pathology appeared to be associated with the sickle-cell trait. Clearly this meant that “the importance of the sickle cell trait appears to be limited to the relatively small grooup who in addition to the trait have sickle cell anemia” (Diggs et al. 1933).
As the distinction between the trait and the anemia became fully accepted, the pace of research on sickle-cell anemia quickened, and in 1949 two milestones were reached almost simultaneously. First, two investigators independently discovered the correct mechanism of inheritance of the trait and the anemia. One of these, a medical officer serving in what was then Rhodesia, compiled several large Bantu pedigrees of the sickle-cell trait. In one he observed two affected parents having a child with sickle-cell anemia. From his studies he concluded that the sickle-cell trait appears as a heterozygote with the normal allele, whereas sickle-cell anemia is the homozygous state of the sickle-cell gene, meaning that it must be inherited from both parents (Beet 1949).
Second, J. V. Neel (1949) reported precisely the same conclusion based on a more statistical analysis. These findings led to the recognition that the vast majority of dominant genetic diseases are heterozygous. The first report on the implications of an altered molecule in the pathogenesis of a human disease should probably be credited to M. Hoerlin and G. Weber, a German physician and a medical student, respectively, who wrote in 1947 that the condition known as methemoglobinemia (14170) was due to a “variant of the globin component of the hemoglobin molecule” (Heller 1969). However, the report that is celebrated is that of L. Pauling and colleagues (1949). Through a set of meticulous experiments they were able to show that the hemoglobin molecules in individuals with sickle-cell anemia and those in normal controls were fundamentally different but, “having found that the electrophoretic mobilities of sickle-cell hemoglobin and normal hemoglobin differ, we are left with the considerable problem of locating the cause of the difference” (Pauling et al. 1949).
They predicted that the abnormality would be found in the globin part of the molecule and not in the heme groups, that the sickling process involved abnormal interactions of altered molecules, and that the alteration resulted in two to four more net positive charges per molecule. Their predictions, we know, were all correct, as was shown by V. M. Ingram in 1956. Denatured sickle-cell hemoglobin (HbS), digested with trypsin, was compared with normal hemoglobin (HbA) after sequential electrophoresis and partition chromatography (Figure III.1.3). The abnormal HbS differed in only one “spot” on the filter, which had a net positive charge. Within three years it was demonstrated that the charge was caused by the alteration of a single amino acid through a mutation (Ingram 1959).
Since the moment the puzzle of sickle-cell anemia was solved, the study of mutant globins and their associated disorders has signaled nearly every major advance in molecular biology.
In 1978 R. M. Lawn and colleagues and Y. W Kan and A. M. Dozy independently demonstrated the first human restriction fragment length polymorphisms (RFLPs) in the region of the beta-chain of the hemoglobin molecule. This led to the explosion of recombinant-DNA-based studies on human genetic diseases as well as human gene mapping (Willard et al. 1985). The discovery of highly repetitive DNA sequences in globin genes has resulted in the development of the hypervariable “minisatellite” and VNTR (variable number of tandem repeat) DNA probes (Jeffreys et al. 1985, 1986; Nakamura et al. 1987). In addition, the use of other recombinant DNA techniques in the molecular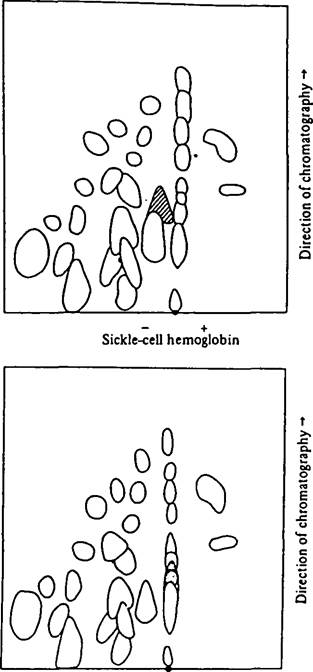
Normal hemoglobin
Figure III.1.3. The original two-dimensional chromatographic spreads of normal and sickle-cell hemoglobins. The different “spot” having a net positive charge is seen to have moved further left in the electrophoretic field, toward the negative pole. The alteration in the pattern represents the substitution of the amino acid valine for glutamic acid. (From Ingram 1956, with permission; copyright 1956 by Macmillan Journals Limited.)
diagnosis of genetic diseases was pioneered with HbS by Kan and Dozy (1978) and is now being applied to a wide range of disorders, including cancer and acquired infectious diseases (Caskey 1987).
The Arrival of Cytogenetics: Down Syndrome
The chromosome theory of heredity, that the Mende- Iian characters were contained in the chromosomes, developed soon after the rediscovery of Mendel’s laws and was in large measure a direct result of the precision of Mendel’s own experimental observations. W. S. Sutton had observed a very high degree of organization of the chromosomes of the orthop- teran genus Brachystola. He concluded that the parallels between the organization and behavior of the chromosomes and the laws of segregation and independent assortment could not be due to chance: “We have reason to believe that there is a definite relation between chromosomes and allelomorphs or unit characters” (Peters 1959).
Sutton proposed that multiple characters would be found on a single chromosome and made a direct reference to genetic linkage: “If then, the chromosomes permanently retain their individuality, it follows that all allelomorphs represented by any one chromosome must be inherited together” (Peters 1959).Spurred by the work of Sutton and others interested in chromosomes, T. H. Morgan and a group of his students at Columbia University began to study the inheritance of unit characters, the individual factors OfMendelian transmission (Allen 1978; Carlson 1981). Using the fruit fly Drosophila as their experimental model, Morgan and his students made a series of fundamental discoveries that led, by 1914, to their conclusion that the Mendelian factors, or genes, were physical entities present in the chromosomes in a linear manner (Sturtevant 1913; Morgan 1914).
In 1923 T. S. Painter suggested that the number of human chromosomes was 48, and this became the accepted number for the next 23 years. Improved cytological techniques eventually led to better resolution of the chromosomes, with the result that J. H. Tjio and A. Levan (1956) established that the correct number of human diploid chromosomes was 2n = 46. So strong had been the belief in Painter’s estimate that, as Tjio and Levan themselves noted, a previous study by another group had been abandoned because “the workers were unable to find all the 48 human chromosomes in their material; as a matter of fact, the number 46 was repeatedly counted in their slides” (Tjio an^ Levan 1956). (Clearly, an open mind is as important as good technique.)
It was not long after the number of human chromosomes was established that the solution to an old and curious medical puzzle became apparent. A condition called furfuraceous idiocy had been described in 1846 by the French physician E. Sdguin. The traits marking the syndrome included characteristic facial features, slow and incomplete growth, and mental retardation. J. Langdon Down ascribed these traits to the Mongol type in 1867: “A very large number of congenital idiots are typical Mongols. So marked is this, that when placed side by side, it is difficult to believe that the specimens are not children of the same parents” (Down 1867).
From this publication the term Mongolian idiot replaced the previous name for the condition, and later the term Down syndrome became interchangeable with it. For the next 92 years repeated studies of Down syndrome failed to produce a viable etiologic hypothesis. L. S. Penrose (1939) summarized the state of knowledge by noting that the disorder had failed to meet Mendelian expectations, though some had favored “irregular dominance” as an explanation, and that the only clear and stable correlate was late maternal age. Penrose (1939) ended his paper by suggesting that “Mongolism and some other malformations may have their origin in chromosome anomalies.” Two decades later the improvements in cytological techniques that allowed the correct number of human diploid chromosomes to be determined also provided the solution to Down syndrome. In 1959 three French cytologists, led by J. Lejeune (1959), announced that patients with Down syndrome had an extra chromosome, which was one of the small telocentric chromosomes later designated as chromosome 21 (Figure III. 1.4).
This first example of a human disorder caused by a specific chromosome anomaly in which there existed at least one triploid chromosome in an otherwise diploid set (now called trisomy) led to a spate of similar findings. These included Turner’s syndrome (caused by the lack of one X-chromosome-XO), Klein- felter’s syndrome (XXY), trisomy 13, and trisomy 18 (Thurman 1986). In 1960 the first international congress was called, in Denver, Colorado, to establish a standardized cytogenetic nomenclature. Much of the impetus came from the discovery of chromosome banding techniques by which each chromosome could be identified individually. Over time these techniques have been refined to the point where subbands and regions within them can be identified (Thurman 1986). However, even with these refinements it has only recently been possible to study directly what specific regions of chromosome 21 are responsible for the Down syndrome phenotype.
As has been the case with sickle-cell anemia and many other human diseases, recombinant DNA technology has enabled researchers to study Down syndrome with greater detail and precision. Chromosome 21 contains less than 2 percent of the DNA in the human genome, yet with an estimated 50,000 genes in the genome (Shows, Sakaguchi, and Naylor 1982), it probably houses 1,000 genes. By means of recombinant DNA and related techniques, more than 20 of those genes, along with nearly 50 anonymous sequences, have been mapped (see Figure ΠI.1.4). Among the mapped genes is one that codes for a ribosomal RNA (RNR-4, 18045). In addition, several genes of medical interest are known. These include the gene for the enzyme cystathionine beta
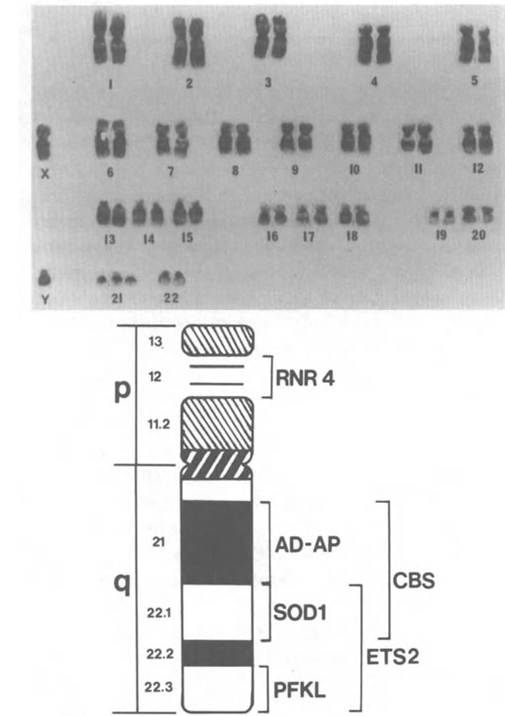
Figure III. 1.4. Top: G-banded chromosome spread of a man with Down syndrome (47, XY, +21). The individual chromosome numbers are designated by convention. Bottom: ideogram of human chromosome 21 indicating the subregional localization of several genes of interest (see text). (From J. A. Fraser-Roberts and Marcus E. Pembrey 1978, with permission.)
synthase (CBS), which is implicated in the recessive metabolic disorder homocysteinuria (23620); the gene for liver-type phosphofructokinase (PFKL), which, in deficiency, leads to a type of hemolytic anemia (17186); and two oncogenes, ETS-2 (16474) and ERG (16508). The precise localization of ETS-2 to band 21q22 was a major indication that this region of chromosome 21 is the one responsible for many of the medical features of Down syndrome.
Individuals with Down syndrome have, in addition to classical facial features and mental retardation, a greatly increased risk of developing myeloid leukemia, a high incidence of cataract, and, in older patients, a neurological degeneration resembling Alzheimer’s disease. The oncogene ETS-2 at 21q22 is known to be rearranged in acute myeloid leukemia (McKusick 1986), and linked to this locus at 21q22 is the locus for the enzyme superoxide dismutase (SOD-1). Y. Groner and colleagues (1986) have reviewed molecular evidence that SOD-I enhances lipid peroxidation, which leads to toxic neurological effects and to cataracts.
The Alzheimer’s disease-like dementia of older Down syndrome patients suggested that the q22 region of chromosome 21 might be the location of the amyloid protein found in the senile plaques of Alzheimer’s patients. D. Goldgaber and colleagues (1987) and R. E. Tanzi and colleagues (1987) independently reported that the amyloid-B protein gene mapped nearby at 21q21. The effect resulting from the extra copy of the amyloid-B protein gene in Down syndrome is an overproduction of amyloid and presenile plaque formation. ι
Finally, the advent of recombinant DNA techniques and sophisticated analytic tools for estimating genetic linkage relationships have enabled one group to focus on the cause of the trisomy. Using several well-mapped anonymous DNA sequences from chromosome 21 as well as a probe for SOD-1, A. C. Warren and colleagues (1987) have demonstrated that recombination among DNA markers on chromosomes 21 that have undergone nondisjunction, the event leading to a trisomy, occurs to a significantly lesser extent than it does in controls. Reduced recombination is indicative of asynapsis, or a failure of normal chromosome pairing. This result will very likely lead to the discovery of the molecular mechanism of chromosome pairing.
A Failure OfExpectations: Kuru
In August 1953, an officer in an Australian government patrol working in the South Fore region of the highlands of Papua New Guinea noted a peculiar condition:
Nearing one of the dwellings [at Amusi], I observed a small girl sitting down beside a fire. She was shivering violently and her head was jerking spasmodically from side to side. I was told that she was a victim of sorcery and would continue thus, shivering and unable to eat, until death claimed her within a few weeks. (J. McArthur 1953, in Lindenbaum 1979)
The condition from which she was suffering was locally called kuru (trembling or fear), a progressive neurological disorder peculiar to that region of Papua New Guinea.
Because the South Fore region was relatively isolated and the villages interrelated through extensive kinship ties, it was thought that kuru was a genetic disease occurring in an isolated district where inbreeding was elevated. However, kuru required that the gene be dominant in women and recessive in men for the epidemiology of the illness to be accounted for by a genetic factor (Bennett, Rhodes, and Robson 1959). Thus, even as early as 1963 the genetic etiology that seemed plausible was being discarded (Lindenbaum 1979).
The case of kuru is mentioned here briefly because it is a good example of the sophistication achieved in medical genetics and the study of human diseases. In the early part of the twentieth century, the power of the Mendelian model was so great that the abandonment of an unwieldy genetic hypothesis, which for kuru took only a couple of years, might have required decades. The true cause of kuru was a bizarre infectious agent that S. B. Prusiner (1982) termed a prion, or proteinaceous infectious particle. In an odd twist, the infectious component in prions, a relatively small protein called the prion protein, PrP 27—30 (Prusiner et al. 1984), was found to have a normal homolog in mammalian genomes, including human ones. PrP produced its effect by forming rodlike structures resembling amyloid, and it was thought that not only was the PrP gene to be found on chromosome 21 but that Alzheimer’s disease and even Down syndrome might be associated with prion infection (Prusiner 1982). However, Y.-C. J. Liao and colleagues (1986) and R. S. Sparkes and colleagues (1986) independently reported cloning the human PrP gene and mapping it to chromosome 20. The story of the prion and of the PrP gene is still unfolding, and although it is not technically a genetic disease, kuru and neurological disorders like it fit well within the multifactorial heuristic model that moved the study of genetic diseases from one of recording oddities to a complex, interdisciplinary enterprise making use of a full range of techniques and technologies.