102 Parkinson’s Disease
Several species of the genus Paragonimus, the lung flukes, can parasitize human beings. The most important, Paragonimus westermani, is found in China, Japan, Korea, Southeast Asia, Papua New Guinea, and parts of India and Central Africa.
It was first discovered in the lungs of tigers in European zoos in 1878. Other species occur in Asia, in Africa, and in Central America and parts of South America. Wild and domestic members of the cat and dog families and other carnivorous animals are also hosts, and in many places humans are accidental hosts for worms that normally reside in other mammals. Adult worms produce eggs in the lungs, which reach fresh water either in the sputum or by being coughed up, swallowed, and passed in the feces.Motile larvae hatch, penetrate an appropriate type of snail, undergo two reproductive cycles, and emerge to seek the second intermediate host, a crab or crayfish. Here they penetrate between the joints of the crustacean’s exoskeleton, and encyst there to await ingestion by humans or other definitive host. They then burrow through the intestinal wall and the diaphragm and enter the lungs, where they may survive for many years. Slow, chronic lung damage may become very serious in heavy infestations. Migrating flukes sometimes wander widely lost and reach atypical (ectopic) sites like the brain, where they cause a variety of neurological symptoms and may prove fatal.
Diagnosis of paragonimiasis depends on detection of the eggs in sputum or feces. Treatment of the lung form of the disease is usually effective, but may be prolonged. Prevention is achieved by avoidance of raw, poorly cooked, pickled, or marinated freshwater crabs and crayfish.
K. David Patterson
Bibliography
Burton, K. 1982. Pulmonary paragonimiasis in Laotian refugee children. Pediatrics 70: 246—8.
Kean, B. H., et al. eds.
1978. Tropical medicine and parasitology: Classic investigations, Vol. II, 601-14. Ithaca and London. [Five early papers.]Nwokolo, C. 1974. Endemic paragonimiasis in Africa. Bulletin of the World Health Organization 50: 569-71.
Yokagama, M. 1965. Paragonimus and paragonimiasis. In Advances in Parasitology, ed. B. Dawes, Vol. II, 99— 158. New York.
Parkinson’s disease, or parkinsonism, is a syndrome (i.e., a constellation of clinical signs and symptoms) consisting of four cardinal features: resting tremor, bradykinesia (physical and mental sluggishness), rigidity, and impaired postural reflexes. The diagnosis is made on the basis of finding any three of the four cardinal features.
Distribution and Incidence
This disease occurs throughout the world, with no population protected against the condition. Most surveys have investigated Caucasian populations of northern European or of Anglo-Saxon descent, and few studies have been done on the occurrence of Parkinson’s disease in other populations. In Caucasians the prevalence is 84 to 187 per 100,000 of population, with no geographic patterns and no clusters of increased incidence. Two studies seem to indicate a lower prevalence in blacks; this has been the clinical experience as well, probably indicating a decreased risk of Parkinson’s disease for blacks. The annual incidence varies from 5 to 24 per 100,000 of the white population. These figures, of course, depend on the methods of ascertainment, the population studied, the length of time that data have been collected, and many other factors. If the prevalence is divided by the annual incidence, the average duration of the illness is approximately 10 years.
Epidemiology
Parkinson’s disease usually occurs in late middle life or beyond. The mean age of onset is 58 to 62. Onset before age 30 is rare but is not unknown, and there is a juvenile form of Parkinson’s disease. The greatest incidence is in the decade age 70 to 79 years, with an incidence of 1 to 2 per 1,000 population per year.
Later the incidence of parkinsonism seems to decline, a finding which, if true, would have important implications about pathogenesis, indicating that the disease is not simply a result of the operation of the aging process on the nervous system. There appears to be no difference between the sexes in regard to the risk of being affected by Parkinson’s disease; early studies in this respect were in error because of decreased ascertainment in females. It is now generally accepted that the ratio is almost or exactly the same for both sexes. Parkinson’s disease was known before 1817, the year of publication of the famous manuscript by James Parkinson, but prevalence studies have been possible only since the 1960s, and they indicate no substantial change in incidence.In 1917 an epidemic of encephalitis Iethargica started in Vienna and spread throughout the world. Following this illness, about half of the victims developed Parkinson’s disease with tremor, bradykinesia, and rigidity often associated with oculogyric crises, parkinsonian crises (sudden episodic worsening of signs and symptoms), behavioral abnormalities, cranial nerve palsies, and a host of other central nervous system abnormalities. The age of onset of the postencephalitic Parkinson’s disease is early compared to other types of Parkinson’s disease. A popular and now discarded theory, the Cohort theory, hypothesized that all Parkinson’s disease was caused by the encephalitis Iethargica agent and that Parkinson’s disease would therefore disappear in the 1980s. Since 1961 when the Cohort theory was promulgated, evidence against it has been overwhelming, and we now know that there is no decrease in the prevalence of Parkinson’s disease, but of course, the postencephalitic Parkinson’s disease has almost disappeared.
Mortality
The mortality rate from parkinsonism varies from 0.5 to 3.8 per 100,000 of population, and the duration averages about 10 years, with a large range of 1 to 33 years, depending on the age of onset, the rate of progression, the patient’s general health, and the treatments received.
M. M. Hoehn and M. D. Yahr found that victims of Parkinson’s disease have a range of mortality 2.9 times greater than the norm, but treatment with Ievodopa increases the quality of life, decreases the symptoms of the disease, and also reduces this excess mortality. Genetic studies failed to show significant risks of parkinsonism in families, and careful analysis of monozygotic twins indicates a lack of concordance for this disease. The twin studies clearly show that the major factor in the cause of parkinsonism is not hereditary.Etiology
The signs and symptoms of parkinsonism are caused by a decrease in striatal dopamine due to the loss of dopaminergic neurons in the substantia nigra of the midbrain. At the present time, environmental agents are the primary known cause of parkinsonism; exposure to manganese, carbon disulfide, and carbon monoxide are recognized environmental toxins that can produce the disorder. It is also known that drugs that interfere with dopaminergic pathways or receptors — such as phenothiazines, reserpine, and alpha-methyldopa - can produce the Parkinson’s syndrome, and multiple head traumas, probably causing atrophy of the substantia nigra, also may do the same. The recent discovery that 1-methyl- 4-phenyl-l,2,3,6-tetrahydropyridine (MPTP), a commercially available chemical intermediate used in the synthesis of organic compounds, induces parkinsonism, has again supported the concept that environmental chemicals play a major role in the pathogenesis of this disorder. Varying amounts of MPTP may be formed as a byproduct in the synthesis of potent analgesic drugs, one of which is MPPP (l-methyl-4-phenyl-4-proprionoxypiperi- dine), the reverse ester of meperidine, a strong analgesic drug similar to heroin and morphine. The selfadministration of small amounts of MPTP by young drug abusers who were using MPPP as an alternative to heroin has resulted in a severe and permanent Parkinson’s syndrome that resembles Parkinson’s disease in its clinical, pathological, and biochemical features as well as its responsiveness to the drugs usually used in the treatment of Parkinson’s patients.
Primate models of parkinsonism using MPTP demonstrate that a Parkinson-Iike syndrome can be produced in the monkey, and it is now known that metabolic conversion by monoamine oxidase B of MPTP to MPP+ (l-methyl-4-phenyl- pyridinium) is the reaction that results in the ultimate toxin MPP+. It therefore follows that blocking monoamine oxidase B with a selective inhibitor such as L-deprenyl completely prevents the toxicity of MPTP, but administration of a selective monoamine oxidase A inhibitor such as clorgylin does not. Thus, MPTP turns out to be the first toxin positively known to cause Parkinson’s disease whose neuropharmacology is known and whose toxicity can be prevented by preventing its metabolic conversion to the ultimate toxin, which is MPP+ (see Figure VIII.102.1).Pathology
Nigrostriatal dopaminergic system failure is the important pathological feature of parkinsonism. Loss of pigment cells in the substantia nigra and the dopaminergic neurons associated with them results in a deficiency of dopa in the striatum and produces the clinical signs and symptoms of the disease.
Clinical Manifestations
The cardinal features of Parkinson’s disease are resting tremor, bradykinesia, rigidity, and postural instability. The resting tremor usually is 4 to 6 cycles per second and is present at rest and decreases with
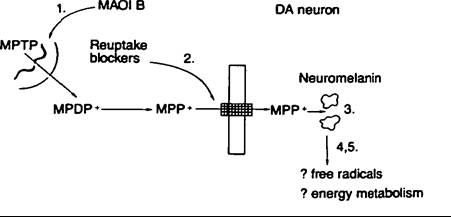
Figure VIII. 102.1. Scheme illustrating the mechanism of MPTP toxicity at nigral dopamine neurons in primates. MPTP, which is lipophilic, enters the brain where it is transformed into MPP+ by monoamine oxidase B located in glial cells or in serotonergic neurons. MPP+ is taken up into the dopaminergic (DA) neurons, where it accumulates, by dopamine reuptake mechanisms. The binding of MPP+ to neuromelanin might assist in the accumulation of MPP+, or might contribute to its toxicity. MPP+ within the neurons is assumed not to undergo redox cycling to reproduce free radical species but, rather, to be actively accumulated by mitochondria, where it inerferes with mitochondrial energy metabolism, leading to cell death. The precise mechanism of MPP+ toxicity remains unknown.
The sequence of events leading to MPTP toxicity can, in theory, be inhibited at a number of points: (1) Selective monoamine oxidase B inhibitors (MAOI B) such as deprenyl inhibit the conversion of MPTP to MPP+; (2) dopamine reuptake blockers such as nomifensine inhibit the entry and accumulation of MPP+ into dopaminergic neurons; (3) chloroquine might inhibit the binding of MPP+ to neuromelanin and thus limit its toxicity for protective substances such as antioxidants (e.g., alpha-tocopherol); or substances such as acetyl-L-carnitine that protect mitochondria from toxic insults might inhibit the toxic mechanism of MPP+; (4,5) other substances as yet unknown might inhibit the production of free radicals or boost energy metabolism. (From Joseph Jankovic and Eduardo Tolosa, eds. 1988. Parkinson’s disease and movement disorders. Baltimore, Maryland: Urban and Schwarzenberg, by permission of the publisher.)action. The tremor usually starts in the upper extremity on one side and then spreads to the lower extremity on the same side. Upper and lower extremities are usually time-locked, with the lower extremity a half cycle behind the upper extremity. Once the tremor has spread to one side of the body, the opposite upper extremity is usually involved, and then the opposite lower extremity. Postural tremor is also present in the majority of Parkinson’s patients, and postural tremor is usually slightly faster than the resting tremor. Bradykinesia, which literally means “slow movement,” is not the same as hypokinesia, meaning “decreased movement,” or akinesia, “no movement.” All of these movement disorders can be part of Parkinson’s disease. The bradykinesia shows up as a delay in the execution and initiation of voluntary movement, and also there is difficulty arresting the movement once it has been started. Akinesia is simply an extreme state of immobility, and some Parkinson’s patients, particularly before treatment with Ievodopa became available, eventually ended their days totally rigid and immobilized. Rigidity is resistance to passive stretch of muscle, and the patients with parkinsonism have a characteristic stiffness of the muscles of the body. This stiffness can be brought out by passive rotation of the wrist or flexion-extension at the forearm. It is also present in the truncal muscles and can be examined by placing one’s hands on the dorsal aspect of the patient’s back muscles and having the patient flex and extend the body at the waist. The rigidity that is characteristic of parkinsonism is the rigidity that usually gives way in little catches and, hence, has been called cogwheel rigidity. The most disabling of the cardinal features of parkinsonism is the postural instability. This one symptom alone accounts for most of the falls that occur in this disease. The instability results in loss of balance with propulsion and retropulsion, and such patients are unable to anticipate a change in their posture and thereby correct for the change, and thus they often follow their center of gravity as they fall to the ground. Little postural corrections that are normally made automatically cannot be done, and this is the major motor defect in the disease. Other clinical features of parkinsonism include masked facies and decreased blinking, increased saliva production called sialorrhea, hyperhidrosis (profuse sweating) or oily skin, a low-pitched monotonous voice, stooped posture, scoliosis, decreased upward gaze, a tendency toward low blood pressure, micrographia, and a shuffling and small-stepped gait.
History
Tremor was mentioned in the writings of Hippocrates, Celsus, and Galen, but the real history of Parkinson’s disease does not start until 1817, the year Parkinson’s essay on the shaking palsy was published - when he was 62 years old. Parkinson called the disease the “shaking palsy,” but he also provided a Latin term: paralysis agitans. He reported on six patients. The first was personally observed, two others were noticed casually in the street; case 4 had an abscess on the chest wall but was lost to follow-up; case 5 was evidently seen at a distance, and there are no details of that patient available; and case 6 was a 72-year-old man who was visually inspected but evidently not examined.
In other words, by modern standards Parkinson’s great essay on Parkinson’s disease would not have passed muster. It is clear that the information he conveys in his essay is basically from visual inspection: He did not examine the patients personally. Despite that his paper was well received by the medical community, and subsequently, clinicians added to his description.
Armand Trousseau, in 1859, included a lecture in clinical medicine on parkinsonism and discussed the rigidity and the bradykinesia. The great French neurologist Jean Martin Charcot considered Parkinson’s disease a neurosis because there was no proper central nervous system lesion to explain it, and the condition was characteristically worsened by stress or emotion. Charcot frequently commented that everything possible had been used to treat parkinsonism but with very little effect. He recommended belladonna alkaloids, especially hyoscyamine, an etropine isomer, now known to us as scopolamine. According to our modern biochemical understanding of parkinsonism, this agent should have a partial effect in improving the signs and symptoms of Parkinson’s disease. In an interesting paper in 1893, Paul Oscar Blocq reported the case of a patient with hemiparkinsonism, who at autopsy was found to have a lesion in the inferior peduncle with complete destruction of the substantia nigra. This, of course, suggested that the neuroanatomic substrate of Parkinson’s disease was the substantia nigra and probably is the first description in the medical literature of a lesion that produces parkinsonism. Many other descriptions were made by William R. Gowers and other famous physicians, but these merely added to the fundamental and original observations of Parkinson.
The road to effective therapy was paved in the 1960s by A. Carlsson, who showed in animals that reserpine produces a bradykinesia that could be reversed by the administration of L-dopa, a dopamine precursor. Because it was known that reserpine administration to humans produces a condition that resembles parkinsonism, L-dopa was administered to reserpinized humans to ameliorate the signs and symptoms of the reserpine-induced parkinsonism. O. Homykiewicz and colleagues then studied the concentration of dopamine in patients dying of parkinsonism, comparing the results to those in normal controls, and reported that there was a marked decrease in dopamine in the caudate nucleus. In 1962 Andre Barbeau tried the oral administration of L- dopa (100 to 200 mg), and in 1961 W. Birkmayer and Hornykiewicz experimented with intravenous levodopa (50 to 150 mg), both reporting temporary benefits in parkinsonism. Others, however, were unable to confirm any major effect until George C. Cotzias and colleagues initiated the modern era of therapy in parkinsonism by showing that much larger oral doses of the L-dopa, 1.6 to 12.6 grams per day, resulted in dramatic improvement in many patients. The improvements were so dramatic that the efficacy of Ievodopa treatment was established beyond any doubt, and a new era in the management of Parkinson’s disease began.
Geography
As was previously mentioned, an increased incidence of Parkinson’s disease has not been shown anywhere in the world. However, there is an increased incidence of a complex of parkinsonism, dementia, and amyotrophic lateral sclerosis (ALS) in Guam, the Kii Peninsula of Japan, Rota, and the western highlands of New Guinea. This peculiar disease, in which patients manifest abnormalities referable to almost every level of the neuraxis, resembles a combination of parkinsonism, Alzheimer’s disease, and ALS. Two major theories have emerged to explain the increased incidence of this condition in areas of the world bordered by the Mariana volcanic fault. The first theory implicates a secondary hyperparathyroidism, which increases the deposition of toxic environmental metals in the nervous system. This theory is supported by the finding of increased manganese and calcium levels in nervous tissue taken from patients dying of this condition. The secondary theory implicates the ingestion of two toxic amino acids - beta-oxaloamine-1- alanine (BOAA) and beta-methylamino-l-alanine (BMAA) - by people in this area of the world. This theory is supported by the finding that infusions of toxic amino acids can produce nervous system disease in primates that does seem to resemble some of the degenerative diseases including parkinsonism, ALS, and Alzheimer’s disease. Further work needs to be done to clarify the role of these toxic amino acids, if any, in the pathogenesis of the Parkinson’s disease- ALS-dementia complex on Guam. Both theories, implicating environmental factors as the cause, are supported by the fact that the incidence of this peculiar combination of disorders is steadily decreasing.
Bernard M. Patten
Bibliography
Barbeau, Andre. 1962. The pathogenesis of Parkinson’s disease. Canadian Medical Association Journal 87: 802-7.
Birkmeyer, W., and O. Homykiewicz. 1961. The l-3,4- Jioxyphenylalanine (DOPA) effect in Parkinson- akinesia. Wiener Klinische Wochenschrift 37: 787-8.
Cotzias, G. C. et al. 1967. Aromatic amino acids and the modification of parkinsonism. New England Journal OfMedicine 276: 347.
Duvoisin, R. C. 1984. Parkinson’s disease: A guide for patient and family. New York.
Duvoisin, R. C. et al. 1963. Parkinsonism before and since the epidemic of encephalitis lethargica. Archives of Neurology 8: 232.
Hoehn, M. M., and M. D. Yahr. 1967. Parkinsonism: Onset, progression, and mortality. Neurology 17: 427.
Homykiewicz, O. 1966. Dopamine (3-hydroxytyramine) and brain function. Pharmacological Reviews 18: 925.
Koller, W. C., ed. 1987. Handbook of Parkinson’s disease. New York.
Parkinson, J. 1817. An essay on the shaking palsy. London.